Adverse Events
Monitoring of adverse events (AEs) is critical to the safety of study animals and data integrity. The Clinical Review Board expects that investigators will assess, document, and review all AEs that occur during a clinical trial at regular intervals.
If serious AEs occur, whether expected or unexpected, these events should be reported to the CRB promptly. Unexpected AEs may require the submission of a protocol amendment to ensure that the informed consent document and clinical study protocol are accurate. How to submit a Reportable Event
The purpose of monitoring AEs includes:
- Identifying events that may have an immediate effect on the safety of the patient
- Informing regulators, investigators, and others of new and important information about events that occur on a clinical trial
- Providing a summary of adverse experiences to develop the drug or regimen toxicity profile
Definitions Associated with Adverse Events
Adverse Event (AE):
COHA vGCP training:
- Any unfavorable and unintended clinical sign or abnormality having been absent at baseline, or, if present at baseline, appears to worsen and is temporally associated with medical treatment or procedure, regardless of the attribution (i.e., relationship of event to medical treatment or procedure)
VIHC GL:
- Any observation in animals that is unfavorable and unintended and occurs after the use of a veterinary product or investigational veterinary product, whether or not considered to be product related.
FDA:
- Any untoward medical occurrence associated with the use of a drug whether or not considered drug-related.
Unexpected Adverse Event (UAE):
FDA:
- An event or reaction that is not listed in the investigator’s brochure or is not listed at the specificity or severity that has been observed; or, if an investigator’s brochure is not required or available, is not consistent with the risk information described in the general investigational plan or elsewhere in the current IND application.
Proposed:
- An event or reaction that is not listed in the informed consent document or clinical study protocol or is not listed at the specificity or severity that has been observed.
Serious Adverse Events (SAE):
COHA vGCP training:
An AE that results in any of the following outcomes, occurring at any dose, is considered serious:
- Death
- Life-threatening consequences
- Inpatient hospitalization or prolongation of existing hospitalization
- Major change in the ability to perform activities or daily living (e.g., eating, sleeping, urinating, defecating)
COHA SAE Reporting:
Any untoward medical occurrence that results in any of the following should be reported:
- Death
- Is life-threatening
- Requires or prolongs hospitalization
- Causes persistent or significant disability/incapacity
- In the opinion of the investigators represents other significant hazards or potentially serious harm to a study subject
A serious adverse event is considered unexpected if it is not described in the clinical study protocols or in the informed consent document.
Reporting Documents and References Regarding Adverse Events
The Clinical Translational Science Award (CTSA) One Health Alliance comprises veterinary schools partnered with medical and other colleagues through a National Institutes of Health Clinical Translational Science Award (CTSA).
COHA’s mission is to advance our understanding of diseases shared by humans and animals. The alliance leverages the expertise of physicians, research scientists, veterinarians, and other professionals to solve medical problems and address the well-being of humans, animals, and the environment.
This approach will capitalize on One Health opportunities that accelerate translational research.

Adverse Event Case Reporting Form
Example form that can be used to capture AEs in a study:
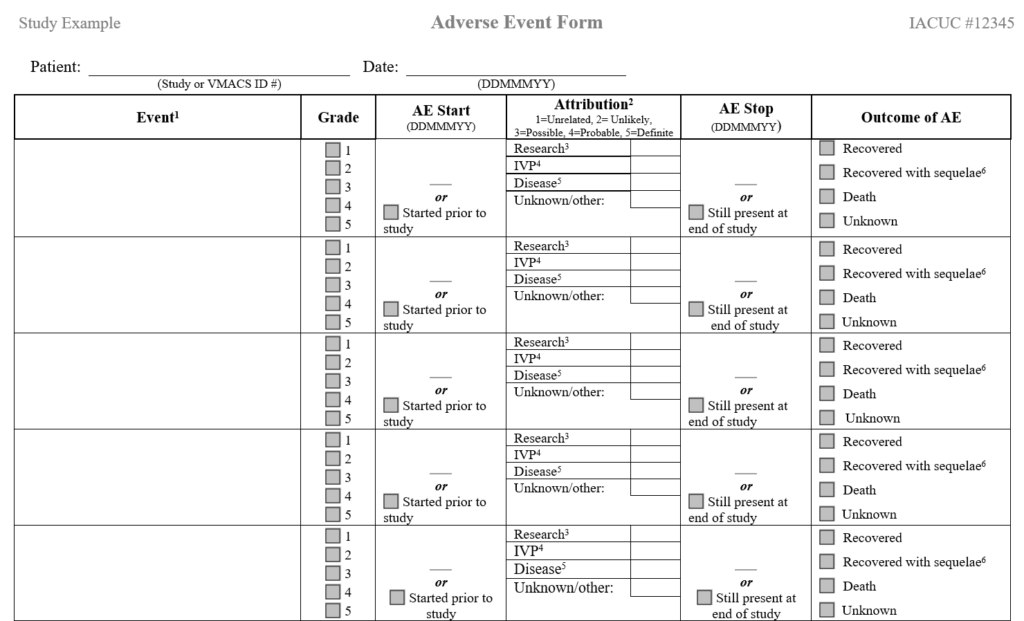
Reference: Veterinary Cooperative Oncology Group
Common Terminology Criteria for Adverse Events (VCOG-CTCAE V2) Following Investigational Therapy in Dogs and Cats
